Generative chemistry with AI has the potential to revolutionize how scientists approach drug discovery and development, health, and materials science and engineering. Instead of manually designing molecules with “chemical intuition” or screening millions of existing chemicals, researchers can train neural networks to propose novel molecular structures tailored to the desired properties. This capability opens up vast chemical spaces that were previously impossible to explore systematically.
While some early successes illustrate the promise that generative AI can accelerate innovation by proposing creative solutions that chemists might not have considered, these triumphs are just the beginning. Generative AI is not yet a magic bullet for molecular design, and translating AI-suggested molecules into the real world is often more difficult than some headlines suggest.
This gap between virtual designs and real-world impact is today’s central challenge in AI-driven molecular design. Computational generative chemistry models need experimental feedback and molecular simulations to confirm that their designed molecules are stable, synthesizable, and functional. Like self-driving cars, AI must be trained and validated with real-world driving data or high-fidelity simulations to navigate unpredictable roads. As illustrated by the new update for the NVIDIA BioNeMo Blueprint for generative virtual screening, without this grounding, both AI systems risk generating promising solutions in theory but fail when put to the test in the physical world.
Oracles: feedback from experiments and high-fidelity simulations
One powerful approach to connecting AI designs with reality is through oracles (also known as scoring functions). In generative molecular design, an oracle is a feedback mechanism—a test or evaluation that tells us how a proposed molecule performs regarding a desired outcome, often a molecular or experimental property (e.g., potency, safety, and feasibility).
This oracle can be:
Experiment-based: an assay that measures how well an AI-designed drug molecule binds to a target protein.
| Experimental? oracle type | Strengths | Limitations | Real-world use |
| In vitro assays (e.g., biochemical, cell-based tests, high-throughput screening) | High biological relevance, fast for small batches, scalable with automation. | Costly, lower throughput than simulations, may not capture in vivo effects. | Standard for identifying and optimizing drug candidates before clinical trials. |
| In vivo models (Animal testing) | Provides insights into safety profiles, dosing, etc., which are often used for drug approval. | Expensive, slow, ethical concerns, species differences may limit relevance to humans. | Used in preclinical drug development, though increasingly supplemented with simulations. |
This oracle is computation-based using high-quality computation (such as molecular dynamic simulations) that accurately predicts a property, such as a free energy method for calculating binding energy (how strongly a drug might fit into an enzyme’s pocket) or a quantum chemistry calculation of a material’s stability. These are in silico stand-ins for experiments when lab testing is slow, costly, or when large-scale evaluation is needed.
| Computational oracle type | Strengths | Limitations | Real-world use |
| Rule-based filters (Lipinski’s Rule of 5, PAINS alerts, etc.) | Quickly flags poor drug candidates, widely accepted heuristics. | Over-simplified, can reject viable drugs. | Used to quickly filter out unsuitable compounds early in drug design. |
| QSAR (Statistical models predicting activity from structure) | Fast, cost-effective, useful for ADMET property screening. | Requires experimental data, struggles with novel chemistries. | Used in lead optimization and filtering out poor candidates. |
| Molecular docking (Structure-based virtual screening) | Rapidly screens large libraries, suggests how molecules bind to targets. | Often inaccurate compared to experimental results, assumes rigid structures. | Common in early drug discovery to shortlist promising compounds. |
| Molecular dynamics & free-energy simulations (Simulating molecule behavior over time) | Models flexibility and interactions more realistically than docking. | Computationally intensive, slow, requires expertise. | Used in late-stage refinement of drug candidates. |
| Quantum chemistry-based methods (First-principles Simulations of electronic structure) | Provides highly accurate predictions of molecular interactions, electronic properties, and reaction mechanisms. | Extremely computationally expensive, scales poorly with system size, and requires significant expertise. | Used for predicting interaction energies, optimizing lead compounds, and understanding reaction mechanisms at the atomic level. |
In practice, researchers often use a tiered strategy where cheap, high-throughput oracles (like quick computational screens) filter the flood of AI-generated molecules. Then, the most promising candidates are evaluated with higher-accuracy oracles (detailed simulations or actual experiments)?. This saves time and resources by focusing expensive lab work on only the top AI suggestions.
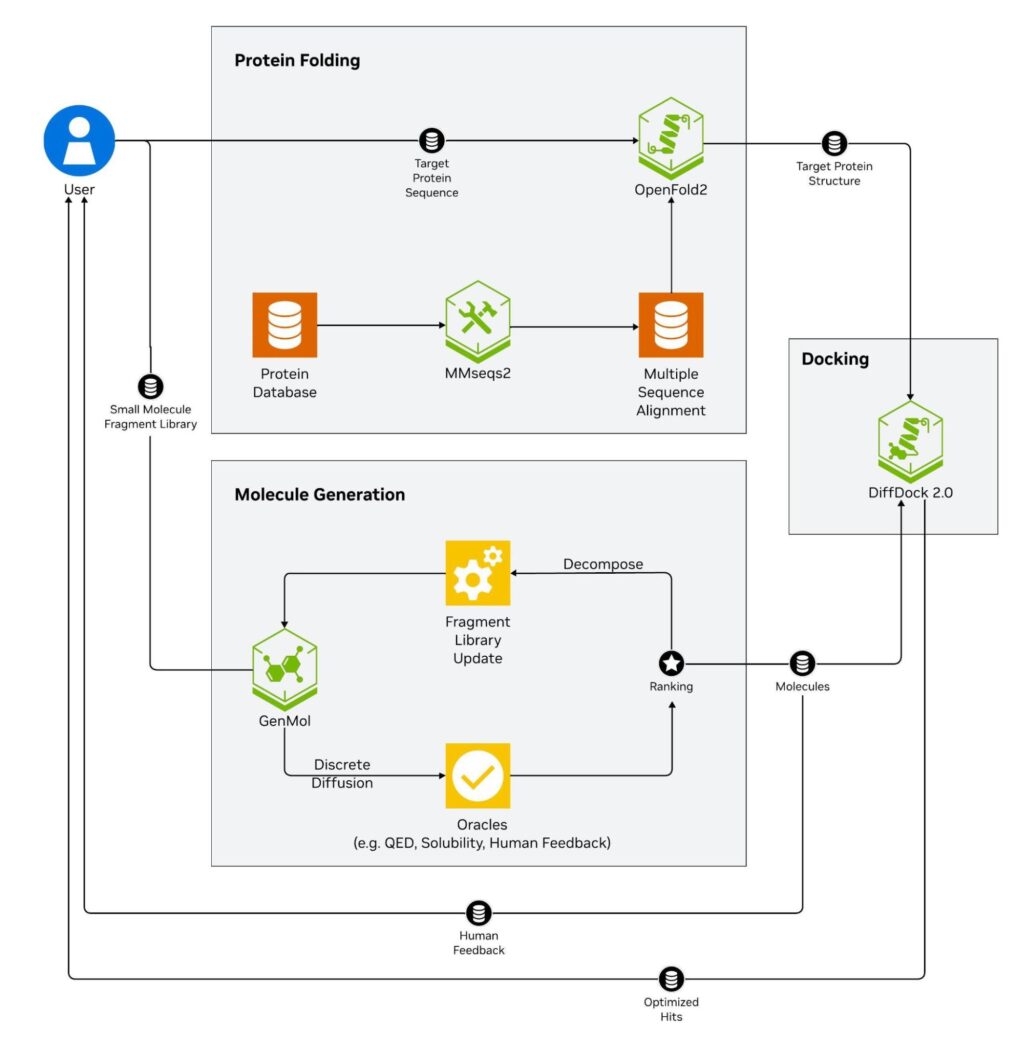
The NVIDIA BioNeMo Blueprint for generative virtual screening (Figure 1) is an example of this:
- The target protein sequence is passed to the OpenFold2 NIM, which accurately determines that protein’s 3D structure using a multiple sequence alignment from the MSA-Search NIM.??
- An initial chemical library is decomposed into fragments and passed to the GenMol NIM, which generates diverse small molecules.
- The initial structures are computationally scored and ranked for multiple characteristics of drugs, such as predicted solubility and Quantitative Estimate of Drug-likeness (QED).
- These scores are used to rank and filter generated molecules during iterative generation cycles until they reach a desirable threshold for further testing. The binding poses for these generated small molecules to the target protein are predicted with the DiffDock NIM.
- Finally, optimized molecules are returned to the user for synthesis and further lab validation.
Oracles in fragment-based molecular design
Follow the pseudocode below to implement an iterative molecule generation and optimization pipeline using NVIDIA GenMol NIM. This process involves generating molecules from a fragment library, evaluating them with an oracle, selecting top candidates, decomposing them into new fragments, and repeating the cycle.
# Import necessary modulesfrom genmol import GenMolModel, SAFEConverter # Hypothetical GenMol APIfrom oracle import evaluate_molecule # Hypothetical oracle functionfrom rdkit import Chemfrom rdkit.Chem import AllChem, BRICSimport random# Define hyperparametersNUM_ITERATIONS = 10 # Number of iterative cyclesNUM_GENERATED = 1000 # Number of molecules generated per iterationTOP_K_SELECTION = 100 # Number of top-ranked molecules to retainSCORE_CUTOFF = -0.8 # Example binding affinity cutoff for filtering# Initialize GenMol modelgenmol_model = GenMolModel()# Load initial fragment library (list of SMILES strings)with open('initial_fragments.smi', 'r') as file: fragment_library = [line.strip() for line in file]# Iterative molecule design loopfor iteration in range(NUM_ITERATIONS): print(f"Iteration {iteration + 1} / {NUM_ITERATIONS}") # Step 1: Generate molecules using GenMol generated_molecules = [] for _ in range(NUM_GENERATED): # Randomly select fragments to form a SAFE sequence selected_fragments = random.sample(fragment_library, k=random.randint(2, 5)) safe_sequence = SAFEConverter.fragments_to_safe(selected_fragments) # Generate a molecule from the SAFE sequence generated_mol = genmol_model.generate_from_safe(safe_sequence) generated_molecules.append(generated_mol) # Step 2: Evaluate molecules using the oracle scored_molecules = [] for mol in generated_molecules: score = evaluate_molecule(mol) # Example: docking score, ML predicted affinity scored_molecules.append((mol, score)) # Step 3: Rank and filter molecules based on oracle scores scored_molecules.sort(key=lambda x: x[1], reverse=True) # Sort by score (higher is better) top_molecules = [mol for mol, score in scored_molecules[:TOP_K_SELECTION] if score >= SCORE_CUTOFF] print(f"Selected {len(top_molecules)} high-scoring molecules for next round.") # Step 4: Decompose top molecules into new fragment library new_fragment_library = set() for mol in top_molecules: # Decompose molecule into BRICS fragments fragments = BRICS.BRICSDecompose(mol) new_fragment_library.update(fragments) # Step 5: Update fragment library for next iteration fragment_library = list(new_fragment_library)print("Iterative molecule design process complete.") |
Oracles in controlled molecular generation
Follow the pseudocode below to implement an iterative, oracle-driven molecular generation process using the MolMIM NIM. This approach involves generating molecules, evaluating them with an oracle, selecting top candidates, and refining the generation process based on oracle feedback (see example code notebook here).
# Import necessary modulesfrom molmim import MolMIMModel, OracleEvaluator # Hypothetical MolMIM and Oracle APIimport random# Define hyperparametersNUM_ITERATIONS = 10 # Number of iterative cyclesNUM_GENERATED = 1000 # Number of molecules generated per iterationTOP_K_SELECTION = 100 # Number of top-ranked molecules to retainSCORE_CUTOFF = 0.8 # Example oracle score cutoff for filtering# Initialize MolMIM model and Oracle evaluatormolmim_model = MolMIMModel()oracle_evaluator = OracleEvaluator()# Iterative molecular design loopfor iteration in range(NUM_ITERATIONS): print(f"Iteration {iteration + 1} / {NUM_ITERATIONS}") # Step 1: Generate molecules using MolMIM generated_molecules = molmim_model.generate_molecules(num_samples=NUM_GENERATED) # Step 2: Evaluate molecules using the oracle scored_molecules = [] for mol in generated_molecules: score = oracle_evaluator.evaluate(mol) # Returns a score between 0 and 1 scored_molecules.append((mol, score)) # Step 3: Rank and filter molecules based on oracle scores scored_molecules.sort(key=lambda x: x[1], reverse=True) # Sort by score (higher is better) top_molecules = [mol for mol, score in scored_molecules[:TOP_K_SELECTION] if score >= SCORE_CUTOFF] print(f"Selected {len(top_molecules)} high-scoring molecules for next round.") # Step 4: Update MolMIM model with top molecules molmim_model.update_model(top_molecules)print("Iterative molecular design process complete.") |
Integrating oracles—experimental and computation-based feedback mechanisms—into AI-driven molecular design fundamentally changes drug design. Researchers can move beyond theoretical molecule generation to practical, synthesizable, and functional drug candidates by establishing a continuous loop between generative models and real-world validation.
This lab-in-the-loop approach enables:
- Faster iteration cycles using AI models like the GenMol NIM and MolMIM NIM to generate and refine molecules based on experimental or high-accuracy computational feedback.
- Efficient resource allocation, where computational oracles quickly screen thousands of molecules before focusing costly lab experiments on the most promising candidates.
- Improved accuracy and generalization by incorporating real-world experimental results into AI models, helping them better predict drug-like properties.
As AI models and oracle systems become more advanced, we’re approaching an era where AI and experimental science co-evolve, driving breakthroughs in drug design. By integrating high-quality oracles, the gap between virtual molecule design and real-world success will continue to shrink, unlocking new possibilities for precision medicine and beyond.
Try oracles for drug design
- Test your own oracles for fragment-based molecule generation in the NVIDIA BioNeMo Blueprint for generative virtual screening.
- Or, swap in the MolMIM NIM for oracle-guided molecular generation.