動態編程( DP )是一種眾所周知的算法技術和數學優化,幾十年來一直被用于解決計算機科學中的突破性問題。
DP 用例的一個示例是使用 Floyd-Warshall 全對最短路徑算法的具有數百或數千個約束或權重的路線優化。另一個用例是使用 Needleman-Wunsch 或 Smith-Waterman 算法進行基因組序列比對的讀取比對。
NVIDIA Hopper GPU 動態編程 X ( DPX )指令加速了基因組學、蛋白質組學和機器人路徑規劃等領域中使用的一大類動態編程算法。加速這些動態編程算法可以幫助研究人員、科學家和從業人員更快地了解潛在的 DNA 或蛋白質結構以及其他幾個領域。
什么是動態編程?
DP 技術最初涉及遞歸地表達算法,其中較大的問題被分解為更容易解決的子問題。
DP 中常用的一種計算優化是保存子問題的結果,并在問題的后續步驟中使用它們,而不是每次都重新計算解決方案。這一步叫做記憶。 Memoization 有助于避免遞歸步驟,而是支持使用迭代查找表–基于配方。先前計算的結果存儲在查找表中。
許多 DP 問題中的一個關鍵觀察結果是,較大問題的解決方案通常涉及計算先前計算的解的最小值或最大值。更大的問題的解決方案在先前解決方案的最小 – 最大值的一個增量內。
一般來說, DP 技術實現了與暴力算法相同的結果,但顯著減少了計算需求和執行時間。
DP 示例:加速 Smith-Waterman 算法
NVIDIA Clara Parabricks 是一套 GPU 加速的基因組分析工具,大量使用 Smith-Waterman 算法,并在 NVIDIA GPU 上運行: A100 、 V100 、 A40 、 A30 、 A10 、 A6000 、 T4 ,以及最新的 H100 。
基因組測序具有廣泛的應用價值,例如個性化藥物或追蹤疾病傳播。生物中的每個細胞都使用 DNA (或堿基)中的四個核苷酸序列來編碼遺傳信息。核苷酸是腺嘌呤、胞嘧啶、鳥嘌呤和胸腺嘧啶,由 A 、 C 、 T 和 G 表示。
像病毒這樣的簡單生物有 10 – 100K 個堿基的序列,而人類 DNA 由大約 30 億個堿基對組成。有一些儀器(基于化學或電信號)對基因物質的短片段的堿基進行排序,稱為讀取。這些讀數通常為 100 – 100K 堿基長,這取決于用于收集讀數的定序器技術。
基因組序列分析中的一個關鍵計算步驟是對齊讀數,以在一對讀數中找到最佳匹配。在第二代測序儀中,這些讀取長度可以是 100-400 個堿基對,在第三代測序儀的讀取長度可以達到 100K 個堿基。對齊讀取是一個重復數千萬或數億次的計算步驟。
在尋找最佳匹配方面存在以下挑戰:
- 基因組中自然發生的變異,使一個物種內的生物具有特定的特征
- 測序儀器或潛在化學過程導致的讀數錯誤
一對讀取之間的最佳匹配相當于一對字符串之間的近似字符串匹配,其中包含獎勵匹配和懲罰差異的步驟。 讀取之間的差異可能是不匹配、插入或刪除。

圖 1 顯示基因組測序中的 Smith-Waterman 步驟旨在找到讀取序列 TGTTACGG 和 GGTTGACTA 之間的最佳匹配。所得到的最佳匹配顯示為 GTT-AC (來自序列 1 ,“ – ”表示刪除)與 GTTGAC (來自于序列 2 )。每個步驟中的得分方案獎勵匹配(+ 3 ),懲罰不匹配( -3 ),并懲罰插入和刪除(參見圖 1 中的差距懲罰公式)。
這是 Smith-Waterman 算法的示例公式。 Smith-Waterman 算法的實現者可以自定義獎勵和懲罰。
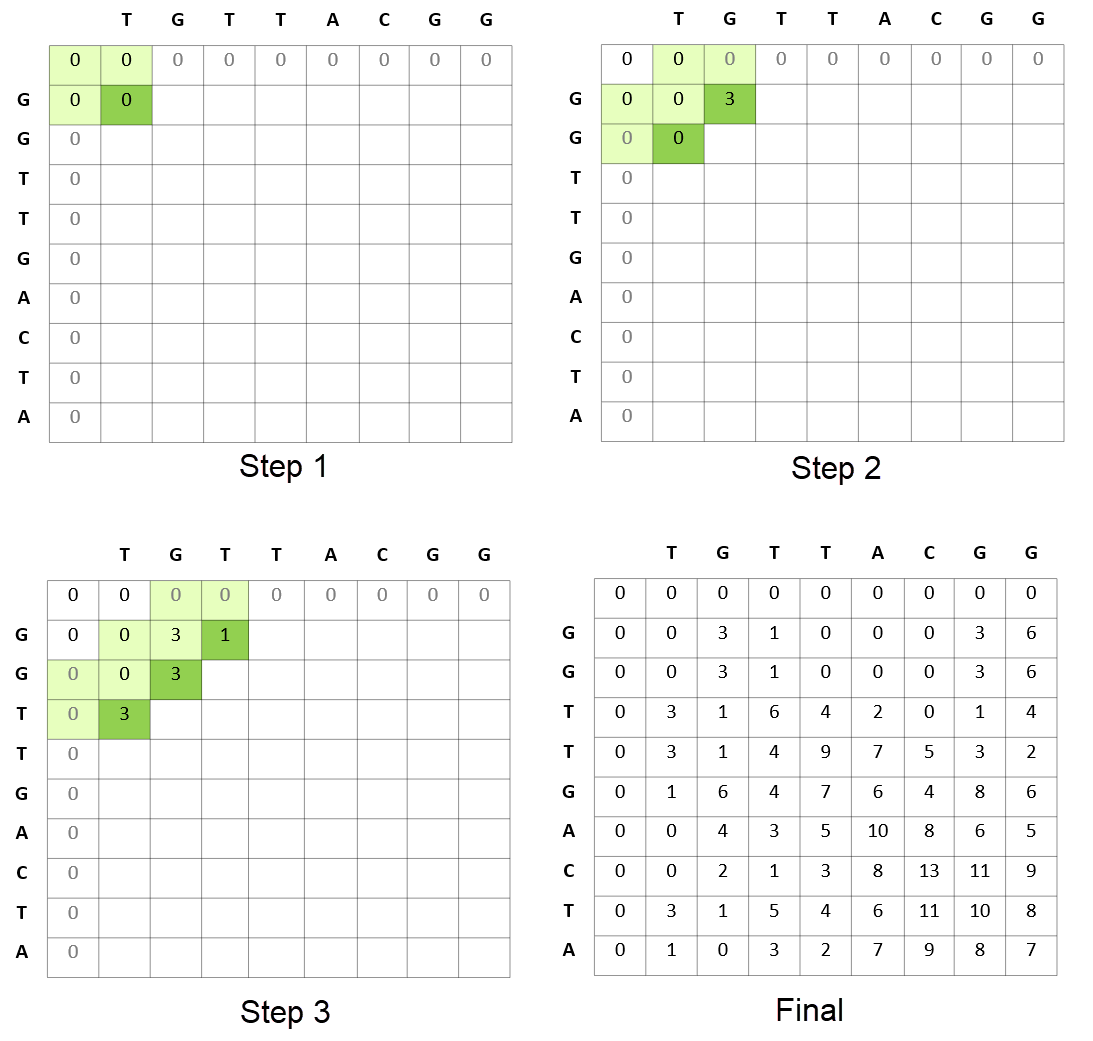
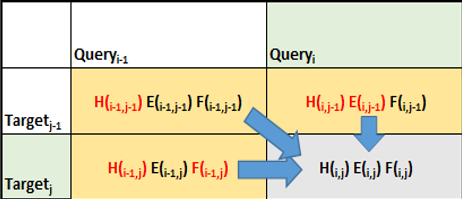
在計算 TGTTACGG 和 GGTTGACTA 之間的最佳匹配的同時, Smith-Waterman 算法還計算 TGTTAC GG 的所有前綴與 GGTTGATA 的所有前綴的最佳匹配。它從這些序列的開頭開始,并使用較小前綴的結果來解決查找較大前綴的解決方案的問題。
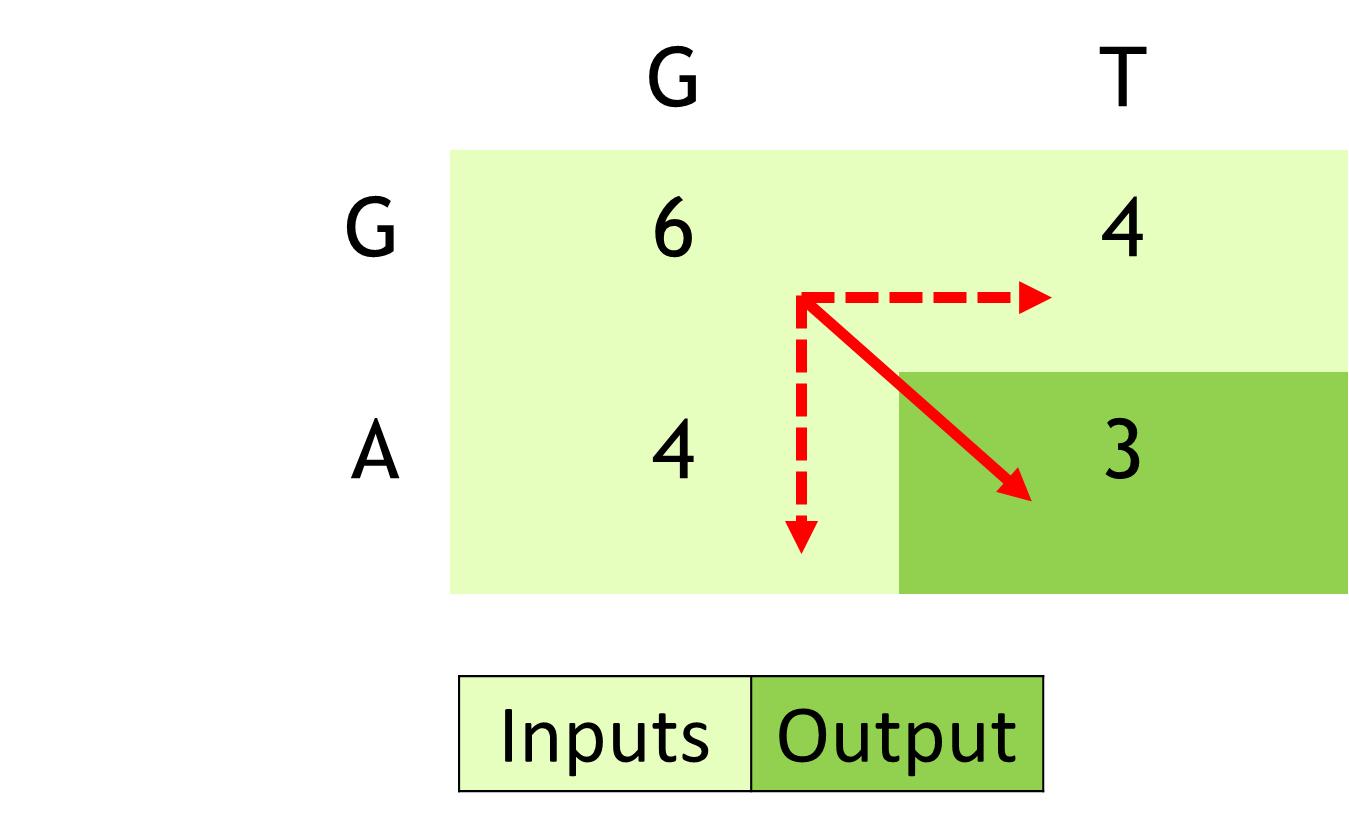
圖 3 顯示了算法如何計算匹配讀取序列的矩陣分數。這種比較匹配是 Smith-Waterman 算法在計算上昂貴的步驟。
這只是 Smith-Waterman 算法如何進行的公式之一。作為示例,不同的公式可以導致算法按行或按列進行。
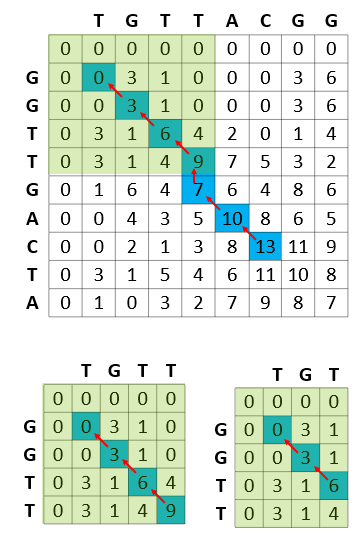
計算得分矩陣后,下一步包括從最高得分回溯到每個得分的原點。這是一個計算簡單的步驟,因為每個單元格都保持其得分的方式(得分計算的源單元格)。
圖 5 顯示了 Smith-Waterman 計算的計算效率,其中算法所解決的每個子問題都存儲在結果矩陣中,并且從不重新計算。
例如,在計算 GGTTGACTA 和 TGTTACGG 的最佳匹配的過程中, Smith-Waterman 算法重用 GGTT ( GGTTGAPTA 的前綴)和 TGTT ( TGTTACGG 的前綴)之間的最佳匹配。反過來,在計算 GGTT 和 TGTT 的最佳匹配時,計算并重用這些字符串的所有前綴的最佳匹配(例如, GGTT 和 TGT 的最佳匹配)。
利用 DPX 指令提高性能
真正的 Smith-Waterman 實現中的內部循環涉及每個單元的以下內容:
- 更新刪除懲罰
- 更新插入懲罰
- 基于更新的插入和刪除懲罰更新分數。

NVIDIA Hopper 架構 math API 為此類計算提供了巨大的加速。 API 將 NVIDIA Hopper 流式多處理器提供的加速作為融合運算(例如,__viaddmin_s16x2_relu,一種按半字執行的內在函數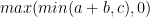
Smith-Waterman 軟件廣泛利用的 API 的另一個示例是三向最小值或最大值,后跟鉗位到零(例如__vimax3_s16x2_relu,一種按半字執行的內在值
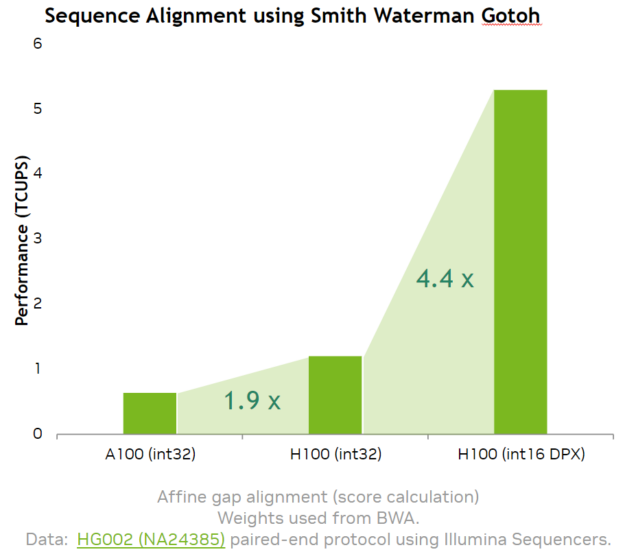
我們使用 NVIDIA Hopper DPX instruction math APIs 實現的 Smith-Waterman 算法的速度比 A100 快 7.8 倍。
Needleman-Wunsch 與部分順序對齊
與 Smith-Waterman 算法使用 DPX 指令的方式相同,有一大類基本上使用相同原理的對齊算法。
示例包括 Needleman-Wunsch algorithm ,其中算法的基本流程與 Smith-Waterman 非常相似。然而,這兩種方法的初始化、插入和間隙懲罰計算不同。
類似 Partial Order Alignment 的算法在其內部循環中密集使用與 Smith-Waterman 單元計算非常相似的單元計算。
所有對最短路徑
具有數千或數萬個對象的機器人路徑規劃是倉庫中的一個常見問題,倉庫中的環境是動態的,有許多移動對象。這些場景可能需要每隔幾毫秒進行一次動態重新規劃。
大多數全對最短路徑算法的內循環如以下 Floyd-Warshall 算法示例所示。偽代碼顯示了全對最短路徑算法如何具有更新每個頂點對之間的最小距離的內循環。最密集的操作基本上是加法運算,然后是最小運算。
initialize(dist); # initialize nearest neighbors to actual distance, all others = infinity
for k in range(V): #order of visiting k values not important, must visit each value
# pick all vertices as source in parallel
Parallel for_each i in range(V):
# Pick all vertices as destinations for the
# above picked source
Parallel for_each j in range(V):
# If vertex k is on the shortest path from
# i to j, then update the value of dist[i][j]
dist[i][j] = min (dist[i][j], dist[i][j] + dist[k][j])
# dist[i][j] calculation can be parallel within each k
# All dist[i][j] for a single ‘k’ must be computed
# before moving to the next ‘k’
Synchronize
DPX 指令所提供的加速功能使其能夠以更少的 GPU 和最佳結果來大幅擴展分析對象的數量或實時進行重新優化。
使用 DPX 指令加速動態編程算法
使用 NVIDIA Hopper DPX 指令, Smith-Waterman 在 A100 GPU 上的速度提高了 7.8 倍,這是許多基因組序列比對和變體調用應用中的關鍵。 CUDA 12 中提供的數學 API 中的公開,使 Smith-Waterman 算法的可配置實現能夠滿足不同的用戶需求,以及 Needleman-Wunsch 等算法。
DPX 指令加速了一大類動態編程算法,如 DNA 或蛋白質測序和機器人路徑規劃。最重要的是,這些算法可以大大加快疾病診斷、藥物發現和機器人自主性,使我們的日常生活變得更好。
致謝
我們要感謝 Bill Dally 、 Alejandro Cachon 、 Mehrzad Samadi 、 Shirish Gadre 、 Daniel Stiffler 、 Rob Van der Wijngaart 、 Joseph Sullivan 、 Nikita Astafev 、 Seamus O ’ Boyle 以及 NVIDIA 的其他許多人。
?