NVIDIA Clara Parabricks v3 的發布。 6 去年夏天,在 全基因組和全外顯子組測序分析綜合工具包 中添加了多個加速體細胞變異調用者和用于注釋和質量控制 VCF 文件的新工具。
在 2022 年 1 月發布的 Clara Parabricks v3 中。 7.NVIDIA 將工具包的范圍擴展到新的數據類型,同時繼續改進現有工具:
- 增加了對 RNASeq 分析的支持
- 通過加速實施 Fulcrum Genomics 的
fgbio管道,增加了對基于 UMI 的基因面板分析的支持 - 增加了對
mutect2正常面板( PON )過濾的支持,使加速mutectcaller符合 GATK 調用腫瘤正常樣本的最佳實踐 - 合并了一個
bam2fq方法,該方法可以加速讀取到新引用的重新對齊 - 使用
ExpansionHunter增加了對短串聯重復分析的支持 - 將呼叫后 VCF 分析步驟加快 15 倍
- 更新了
HaplotypeCaller以匹配 GATK v4 。 1 ,并將DeepVariant更新為 v1 。 1
Clara Parabricks v3 。 7 顯著拓寬了 Clara Parabricks 的功能范圍,同時繼續投資于領先的全基因組和全外顯子組管道領域。
使參考基因組與 bam2fq 和 fq2bam 重新對齊
為了解決人類參考基因組的最新更新問題,并使重新排列讀數便于大型研究, NVIDIA 開發了一種新的bam2fq工具。 Parabricks bam2fq可以從 BAM 文件中提取 FASTQ 格式的讀取數據,為 GATK SamToFastq或bazam等工具提供了一個加速的替代品。
與 Parabricks fq2bam相結合,您可以使用八個 NVIDIA V100 GPU 在 90 分鐘內將一個 30 倍 BAM 文件從一個引用(例如 hg19 )完全重新對齊到一個更新的引用( hg38 或 CHM13 )。內部基準測試表明,與僅依賴 hg19 相比,重新調整到 hg38 并重新運行變體調用可以在一瓶 HG002 真值集中捕獲基因組中數千個真正的陽性變體。
重新調整后的變體調用的改進幾乎與最初與 hg38 一致。雖然這個工作流程以前是可行的,但它的速度非常慢。 NVIDIA 最終將參考基因組更新應用于 Clara Parabricks 中最大的 WGS 研究。
| ############# | |
| ## Download the 30X hg19-aligned bam from Google's public sequencing of HG002 | |
| ## and the respective BAI file. | |
| ############# | |
| wget https://storage.googleapis.com/brain-genomics-public/research/sequencing/grch37/bam/hiseqx/wgs_pcr_free/30x/HG002.hiseqx.pcr-free.30x.dedup.grch37.bam | |
| wget https://storage.googleapis.com/brain-genomics-public/research/sequencing/grch37/bam/hiseqx/wgs_pcr_free/30x/HG002.hiseqx.pcr-free.30x.dedup.grch37.bam.bai | |
| ############# | |
| ## Prepare the references so we can realign reads | |
| ############# | |
| ## Download the original hg19 / hsd37d5 reference | |
| ## and create and FAI index | |
| wget ftp://ftp-trace.ncbi.nih.gov/1000genomes/ftp/technical/reference/phase2_reference_assembly_sequence/hs37d5.fa.gz | |
| gunzip hs37d5.fa.gz | |
| samtools faidx hs37d5.fa | |
| ## Download GRCh38 | |
| wget ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/000/001/405/GCA_000001405.15_GRCh38/seqs_for_alignment_pipelines.ucsc_ids/GCA_000001405.15_GRCh38_no_alt_analysis_set.fna.gz | |
| gunzip GCA_000001405.15_GRCh38_no_alt_analysis_set.fna.gz | |
| ## Make a .fai index using samtools faidx | |
| samtools faidx GCA_000001405.15_GRCh38_no_alt_analysis_set.fna | |
| ## Create the BWA indices | |
| bwa index GCA_000001405.15_GRCh38_no_alt_analysis_set.fna | |
| ## Download the Gold Standard indels from 1kg to use as your known-sites file. | |
| wget https://storage.googleapis.com/genomics-public-data/resources/broad/hg38/v0/Mills_and_1000G_gold_standard.indels.hg38.vcf.gz | |
| ## Also grab the tabix index for the file | |
| wget https://storage.googleapis.com/genomics-public-data/resources/broad/hg38/v0/Mills_and_1000G_gold_standard.indels.hg38.vcf.gz.tbi | |
| ############ | |
| ## Run the bam2fq tool to extract reads from the BAM file | |
| ## Adjust the --num-threads argument to reflect the number of cores on your system. | |
| ## With 8 GPUs and 64 vCPUs this should take ~45 minutes. | |
| ############ | |
| time pbrun bam2fq \ | |
| --ref hs37d5.fa \ | |
| --in-bam HG002.hiseqx.pcr-free.30x.dedup.grch37.bam \ | |
| --out-prefix HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq \ | |
| --num-threads 64 | |
| ############## | |
| ## Run the fq2bam tool to align reads to GRCh38 | |
| ############## | |
| time pbrun fq2bam \ | |
| --in-fq HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq_1.fastq.gz HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq_1.fastq.gz \ | |
| --ref Homo_sapiens_assembly38.fasta \ | |
| --knownSites Mills_and_1000G_gold_standard.indels.hg38.vcf.gz \ | |
| --out-bam HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq.hg38.bam \ | |
| --out-recal-file HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq.hg38.BQSR-REPORT.txt |
 by
by RNASeq 轉錄本定量和融合調用 Clara Parabricks 的更多選項
在 3.7 版中, Clara Parabricks 還添加了兩個用于 RNASeq 分析的新工具。
轉錄本定量是對 RNASeq 數據進行的最有效的分析之一。 Kallisto 是一種基于偽比對的快速表達量化方法。雖然 Clara Parabricks 已經將 STAR 納入了 RNASeq 比對,但 Kallisto 添加了一種補充方法,可以運行得更快。
融合調用是另一種常見的 RNASeq 分析。在 Clara Parabricks 3.7 中, Arriba 提供了第二種方法,用于根據星形對齊器的輸出調用基因融合。與恒星聚變相比,阿里巴可以調用更多類型的事件,包括:
- 病毒整合位點
- 內部串聯復制
- 全外顯子重復
- 環狀 RNA
- 涉及免疫球蛋白和 T 細胞受體位點的增強子劫持事件
- 內含子和基因間區域中的斷點
Kallisto 和 Arriba 的加入使 Clara Parabricks 成為許多轉錄組分析的綜合工具包。
簡化和加速基因面板和 UMI 分析
雖然全基因組和全外顯子組測序在研究和臨床實踐中越來越普遍,但基因面板在臨床領域占據主導地位。
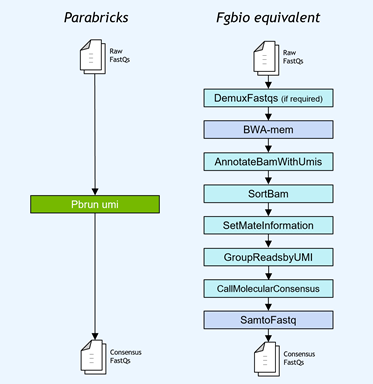
基因小組工作流程通常使用獨特的分子標識符( UMI )連接到讀取,以提高低頻突變的檢測極限。NVIDIA 加速了 Fulcrum Genomics fgbio UMI 管道,并將八步管道整合到 v3 中的單個命令中。 7 ,支持多種 UMI 格式。
使用 ExpansionHunter 檢測短串聯重復序列中的變化
短串聯重復序列( STR )是某些神經系統疾病的公認原因,也是法醫學和群體遺傳學研究中指紋樣本的重要標記。
NVIDIA 通過在 3.7 版中添加對ExpansionHunter的支持,在 Clara Parabricks 中實現了這些位點的基因分型。現在完全使用 Clara Parabricks 命令行界面就可以輕松地從原始讀取轉換為基因型 STR 。
利用 PON 支持改善靜音體細胞突變通話
根據已知正常樣本中的一組突變篩選體細胞突變調用是一種常見做法,也稱為正常組( PON )。 NVIDIA 在mutectcaller工具中增加了對公共 PON 集和自定義 PON 的支持,該工具現在為體細胞突變呼叫提供了 GATK 最佳實踐的加速版本。
加速呼叫后 VCF 注釋和質量控制
在 v3 中。在第 6 版中, NVIDIA 添加了vbvm、vcfanno、frequencyfiltration、vcfqc和vcfqcbybam工具,使呼叫后 VCF 合并、注釋、過濾、過濾和質量控制更易于使用。
v3 。 7 版本通過完全重寫vbvm、vcfqc和vcfqcbybam的后端對這些工具進行了改進,所有這些工具現在都更加健壯,速度提高了 15 倍。
在vcfanno的案例中, NVIDIA 開發了一個名為snpswift的新注釋工具,它帶來了更多功能和加速,同時保留了 VCF 文件精確等位基因數據庫注釋的基本功能。新的snpswift工具還支持用 ENSEMBL 的基因名數據注釋 VCF 文件,有助于理解編碼變體。而新的 post 調用管道看起來與 v3 中的類似。 6 .你會發現你的分析速度更快。
| #!/bin/bash | |
| ######################## | |
| ## In this gist, we'll reuse the commands from our 3.6 tutorial to align reads and generate BAM files. | |
| ## Check out the full post at https://medium.com/@johnnyisraeli/accelerating-germline-and-somatic-genomic-analysis-of-whole-genomes-and-exomes-with-nvidia-clara-e3deeae2acc9 | |
| and Gists at: | |
| ## https://gist.github.com/edawson/e84b2785db75d3c0aea9cc6a59969d45#file-full_pipeline_and_data_prep_parabricks3-6-sh | |
| ## and | |
| ## https://gist.github.com/edawson/e84b2785db75d3c0aea9cc6a59969d45#file-step_1_align_reads_parabricks3-6-sh | |
| ########### | |
| ########### | |
| ## We'll run this tutorial on a GCP VM with 64 vCPUs, 240GB of RAM, and 8x NVIDIA V100 GPUs | |
| ## To save costs, you can also run this on a GCP VM with 32 vCPUS, 120GB of RAM, and 4x V100 GPUs | |
| ########### | |
| ## After aligning our reads, we'll rerun the variant calling stages of our past gist | |
| ## since we've updated the haplotypecaller and DeepVariant tools. We'll | |
| ## also run Strelka2 as an additional variant caller. | |
| ## | |
| ## After that, we'll merge our VCFs to generate a union callset and an intersection VCF | |
| ## with variants called by all three variant callers, annotate our new intersection VCF, | |
| ## and remove variants that fail certain criteria for population frequency. | |
| ## Finally, we'll run our vcfqc and vcfqcbybam tools to generate simple quality control reports. | |
| ############# | |
| ################ | |
| ## HaplotypeCaller | |
| ## This step should take roughly 15 minutes on our 8xV100 VM. | |
| ################ | |
| time pbrun haplotypecaller \ | |
| --ref ~/refs/Homo_sapiens_assembly38.fasta \ | |
| --in-bam HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq.hg38.bam \ | |
| --in-recal-file HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq.hg38.BQSR-REPORT.txt \ | |
| --out-variants HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq.hg38.pb.haplotypecaller.vcf | |
| ################ | |
| ## DeepVariant | |
| ## This step should take approximately 20 minutes on an 8xV100 VM | |
| ################ | |
| time pbrun deepvariant \ | |
| --ref Homo_sapiens_assembly38.fasta \ | |
| --in-bam HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq.hg38.bam \ | |
| --out-variants HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq.hg38.pb.deepvariant.vcf | |
| ############### | |
| ## Strelka | |
| ## This step should take ~10 minutes on a 64-core VM. | |
| ############### | |
| time pbrun strelka \ | |
| --ref Homo_sapiens_assembly38.fasta \ | |
| --in-bams HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq.hg38.bam \ | |
| --out-prefix HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq.hg38.pb.strelka \ | |
| --num-threads 64 | |
| ## Copy strelka results to current directory. | |
| cp HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq.hg38.pb.strelka.strelka_work/results/variants/variants.vcf.gz* . | |
| ## BGZIP and tabix-index the deepvariant VCFs | |
| bgzip -@16 HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq.hg38.pb.deepvariant.vcf | |
| tabix HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq.hg38.pb.deepvariant.vcf.gz | |
| ## BGZIP and tabix index the haplotypecaller VCFs | |
| bgzip -@16 HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq.hg38.pb.haplotypecaller.vcf | |
| tabix HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq.hg38.pb.haplotypecaller.vcf.gz | |
| ## Run the votebasedvcfmerger tool to generate a union and intersection VCF. | |
| time pbrun votebasedvcfmerger \ | |
| --in-vcf strelka:variants.vcf.gz \ | |
| --in-vcf deepvariant:HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq.hg38.pb.deepvariant.vcf.gz \ | |
| --in-vcf haplotypecaller:HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq.hg38.pb.haplotypecaller.vcf.gz \ | |
| --min-votes 3 | |
| --out-dir HG002.realign.vbvm | |
| ## The HG002.realign.vbvm directory should now contain a | |
| ## unionVCF.vcf file with the union callset of HaplotypeCaller, Strelka, and DeepVariant | |
| ## and aa filteredVCF.vcf file with only calls produced by all three callers. | |
| ## Annotate the intersection VCF with gnomAD, ClinVar, 1000 Genomes | |
| ## Download our annotation VCFs and tabix indices | |
| wget https://ftp.ncbi.nlm.nih.gov/pub/clinvar/vcf_GRCh38/clinvar.vcf.gz | |
| wget https://ftp.ncbi.nlm.nih.gov/pub/clinvar/vcf_GRCh38/clinvar.vcf.gz.tbi | |
| wget https://storage.googleapis.com/gcp-public-data--gnomad/release/2.1.1/liftover_grch38/vcf/exomes/gnomad.exomes.r2.1.1.sites.liftover_grch38.vcf.bgz | |
| wget https://storage.googleapis.com/gcp-public-data--gnomad/release/2.1.1/liftover_grch38/vcf/exomes/gnomad.exomes.r2.1.1.sites.liftover_grch38.vcf.bgz.tbi | |
| wget http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/1000_genomes_project/release/20181203_biallelic_SNV/ALL.wgs.shapeit2_integrated_v1a.GRCh38.20181129.sites.vcf.gz | |
| wget http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/1000_genomes_project/release/20181203_biallelic_SNV/ALL.wgs.shapeit2_integrated_v1a.GRCh38.20181129.sites.vcf.gz.tbi | |
| ## Download an Ensembl GTF to annotate the VCF file with gene names | |
| wget http://ftp.ensembl.org/pub/release-105/gtf/homo_sapiens/Homo_sapiens.GRCh38.105.gtf.gz | |
| ## Unzip the GTF file and add the "chr" prefix to the chromosome names (Ensembl excludes this prefix by default. | |
| gunzip Homo_sapiens.GRCh38.105.gtf.gz | |
| awk '{if($0 !~ /^#/) print "chr"$0; else print $0}' Homo_sapiens.GRCh38.105.gtf > Homo_sapiens.GRCh38.105.chr.gtf | |
| time pbrun snpswift \ | |
| --input-vcf HG002.realign.vbvm/filteredVCF.vcf \ | |
| --anno-vcf 1000Genomes:ALL.wgs.shapeit2_integrated_v1a.GRCh38.20181129.sites.vcf.gz \ | |
| --anno-vcf gnomad_v2.1.1:gnomad.exomes.r2.1.1.sites.liftover_grch38.vcf.bgz \ | |
| --anno-vcf ClinVar:clinvar.vcf.gz \ | |
| --ensembl Homo_sapiens.GRCh38.105.chr.gtf \ | |
| --output-vcf HG002.realign.3callers.annotated.vcf | |
| ################## | |
| ## frequencyfiltration | |
| ## Next we'll filter our VCF to remove variants with 1000Genomes allele frequency > 0.05 | |
| ## and gnomAD AF < 0.05 | |
| ################## | |
| time pbrun frequencyfiltration \ | |
| --in-vcf HG002.realign.3callers.annotated.vcf \ | |
| --and-expression "1000Genomes_AF < 0.05" \ | |
| --and-expression "gnomad_v2.1.1_AF < 0.05" \ | |
| --out-vcf HG002.realign.3callers.annotated.filtered.vcf | |
| ################## | |
| ## Finally, we'll run our automated vcfqc tool to generate some | |
| ## basic QC stats. The vcfqcbybam tool could also be run | |
| ## to produce QC stats using an auxilliary BAM file (e.g., when variant calls don't have the desired fields). | |
| ################## | |
| time pbrun vcfqc --in-vcf HG002.realign.3callers.annotated.filtered.vcf \ | |
| --output-dir HG002.realign.3callers.annotated.filtered.qc \ | |
| --depth haplotypecaller_DP --allele-depth deepvariant_AD |
總結
帶有 Clara Parabricks v3 。 7 .NVIDIA 致力于使 Parabricks 成為加速基因組數據分析的最全面解決方案。它是 WGS 、 WES 和現在的 RNASeq 分析以及基因面板和 UMI 數據的廣泛工具包。
有關 3.7 版的更多信息,請參閱以下參考資料:
測試 Clara Parabricks 免費 90 天 并根據自己的數據運行本教程。