GROMACS 是一種廣泛用于模擬生物分子系統的科學軟件包,在理解對疾病預防和治療重要的重要生物學過程中發揮著至關重要的作用。 GROMACS 可以并行使用多個 GPU 以盡可能快地運行每個模擬。
在過去幾年中, NVIDIA 和 主要 GROMACS 開發人員?合作進行了一系列多 GPU 和多節點優化。
在這篇文章中,我們展示了這些改進中的最新進展,通過啟用 GPU 粒子網格 Ewald ( PME )分解和 GPU directcommunication :新 GROMACS 2023 發布版本中提供的一項功能。我們觀察到,通過這項工作,性能提高了 21 倍。
實現改進的多節點性能
在 之前的文章?中,我們對單個節點內的多 GPU 可伸縮性進行了優化,包括 GPU direct 通信的開發。我們描述了 GROMACS 通常如何將一個 GPU 分配給 PME 長程力計算(通過到傅里葉空間的變換執行),而剩余的 GPU 用于短程粒子 – 粒子( PP )力計算(直接在真實空間中執行)。
我們還描述了直接在這些 GPU 之間進行通信的發展:多個 PP GPU + PP-PME 通信之間的光環交換。我們在論文 Heterogeneous parallelization and acceleration of molecular dynamics simulations in GROMACS 中更詳細地描述了這項工作。
GPU 與 MPI 直接通信
在 GROMACS 2022 發布版本中,擴展了 GPU 直接通信功能,以支持與 CUDA 感知 MPI 的兼容性,從而實現跨多個節點(以及每個節點內)的 GPU 直接通信。有關如何激活此功能的更多信息,請參閱本文后面的 How to run 部分。
然而,仍然存在與 PME 的單個 GPU 的限制相關的可擴展性限制。雖然可以增加更多 GPU 來處理 PP 力計算,但不可能將大多數模擬擴展到幾個節點之外。當放大時,單個 PME GPU 在某一點上成為限制因素。 PME 計算必須在多個 GPU 中進行分解,以實現進一步的縮放。
PME GPU 分解
通過引入 PME GPU 分解,在全新 GROMACS 2023 發布版本中,這一單一 PME GPU 限制已被解除。這利用了新的 NVIDIA cuFFTMp library ,該庫能夠以分布式方式跨計算節點內和跨計算節點的多個 GPU 執行所需的快速傅里葉變換( FFT )。
cuFFTMp 則使用 NVSHMEM ,這是一種并行編程接口,可實現快速的單邊通信。它可以有效地利用節點內和節點間互連來執行分布式 3D FFT 所需的所有通信。
通過集成這一新功能, GROMACS 現在可以在模擬中計算多個 PME 等級,從而顯著增強了可擴展性和性能。為了便于移植,該實現還支持 heFFTe ,這是 cuFFTMp 的替代方案。我們發現 cuFFTMp 的速度快了 2 倍,因此我們將在本篇文章的剩余部分重點關注這一點。
這項工作涉及到對 GROMACS 中處理 PME 通信的方式的幾個復雜調整,以及與新 cuFFTMp 功能的接口。
為了實現這一新功能,進行了一些算法改進。這包括引入流水線以增強計算和通信的重疊,以及重新設計并行實現以使用網格重疊減少機制,而不是在 PME 計算之前重新分配數據。
有關更多信息,請參閱 GROMACS GitLab 問題 Support PME decomposition in CUDA backend 和 Optimize CUDA PME Decomposition 。
績效結果
在本節中,我們將展示使用新性能特性的性能結果。我們使用了以下測試用例:
- 衛星煙草花葉病毒( STMV )具有約 1M 個原子,描述于 Heterogeneous parallelization and acceleration of molecular dynamics simulations in GROMACS 。可從 Supplementary Information for Heterogeneous Parallelization and Acceleration of Molecular Dynamics Simulations in GROMACS 獲得。
- BenchPEP-h 具有約 12M 個原子,可從 Max Planck GROMACS Benchmark Suite. 獲得
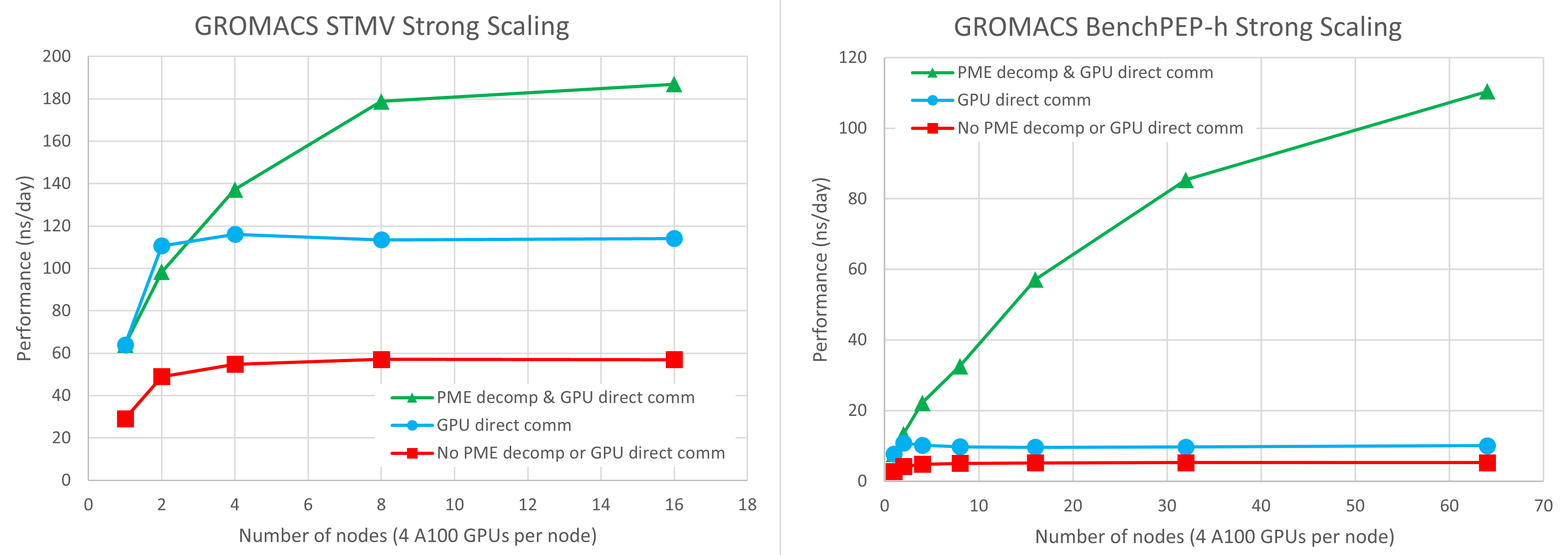
在圖 1 中,我們在 Selene DGX-A100 集群上每個節點使用了四個 NVIDIA A100 GPU 。有關更多信息,請參閱本文的下一節。紅色方塊、藍色圓圈和綠色三角形分別表示使用傳統代碼路徑、僅 GPU 直接通信和結合 GPU PME 分解的 GPU 直接通信的結果。
您可以看到, GPU 直接通信的引入比通過 CPU 內存進行通信的傳統代碼路徑的速度提高了 2-3 倍。通過 GPU 直接通信,基準可以擴展到大約兩個節點,但在這一點上,單個 PME GPU 成為限制因素,不可能進一步擴展。
結合 GPU 直接通信和 PME GPU 分解的結果使用每個節點一個 PME GPU ,我們發現這給出了最佳平衡。每個節點上的剩余三個 GPU 專用于 PP 短程力計算。
在單個節點上, PME GPU 分解沒有影響,因為您仍然只有一個 PME GPU 。
在兩個節點上,使用兩個 PME GPU 的性能與使用單個 PME GPU 的性能類似。前者對于 STMV 稍慢,對于 benchPEP-h 稍快。
在這種規模下,關鍵路徑仍然對 PP 工作負載敏感,因此需要進行實驗以找到性能最佳的配置。例如,我們犧牲了一個 PP GPU 來啟用額外的 PME GPU 。然而,隨著節點數量的進一步擴大,關鍵路徑完全由 PME 性能決定, PME GPU 分解實現了更好的可伸縮性。
STMV 案例可以擴展到八個節點,在這種情況下,結果比傳統代碼路徑快 3 倍,比沒有 GPU PME 分解的結果快 1.6 倍。
對于 STMV ,在 16 個節點上,您可以看到比 8 個節點稍有改進,但并行效率要低得多。較大的benchPEP-h案例可以擴展到測試的最大 64 節點( 256-GPU )配置,比傳統代碼路徑快 21 倍,比沒有 GPU PME 分解的結果快 11 倍。
如何構建和運行 GROMACS
在本節中,我們將詳細介紹如何使用 PME GPU 分解運行 GROMACS 以獲得性能結果。
我們按照網站上的下載和安裝說明安裝了 NVIDIA HPC SDK 22.11 。對于 GROMACS 2023 ,我們不推薦任何更高版本的 HPC SDK ,因為兼容性問題將在未來的開發中解決。
獲取 GROMACS 2023 發布版本如下:
git clone https://gitlab.com/gromacs/gromacs.git cd gromacs git checkout v2023
按如下方式構建 GROMACS :
# make a new directory to build in
mkdir build
cd build
# set the location of the math_libs directory in the NVIDIA HPC installation
HPCSDK_LIBDIR=/lustre/fsw/devtech/hpc-devtech/alang/packages/nvhpc/nvhpc_2022_2211_Linux_x86_64_cuda_11.8-install/Linux_x86_64/2022/math_libs
# build the code with PME GPU decomposition with cuFFTMp enabled,
# in an environment with a CUDA-aware OpenMPI installation
# (see https://manual.gromacs.org/current/install-guide/index.html)
cmake \
../ \
-DGMX_OPENMP=ON -DGMX_MPI=ON -DGMX_BUILD_OWN_FFTW=ON \
-DGMX_GPU=CUDA -DCMAKE_BUILD_TYPE=Release -DGMX_DOUBLE=off \
-DGMX_USE_CUFFTMP=ON -DcuFFTMp_ROOT=$HPCSDK_LIBDIR
make -j 8 #Build using 8 CPU threads. Can increase this if you have more CPU cores available.
HPCSDK_LIBDIR變量設置為 HPC SDK 安裝的Linux_x86_64/2022/math_libs子目錄。
獲取輸入文件如下:
wget https://zenodo.org/record/3893789/files/GROMACS_heterogeneous_parallelization_benchmark_info_and_systems_JCP.tar.gz tar zxvf GROMACS_heterogeneous_parallelization_benchmark_info_and_systems_JCP.tar.gz ln -s GROMACS_heterogeneous_parallelization_benchmark_info_and_systems_JCP/stmv/topol.tpr .
我們在 NVIDIA Selene 集群上進行了基準測試,該集群由多個 DGX-A100 服務器組成,每個服務器都有八個 A100-SXM4 GPU ,通過 Infiniband 連接。然而,根據更典型的 HPC 安裝,每個節點只使用了四個 GPU 。
我們在將 STMV topol.tpr輸入文件復制到工作目錄后,使用標準作業調度方法(本例中為 Slurm )運行代碼。我們提供了 full submission script used on Selene ,其中包括詳細的評論。
要使用 PME GPU 分解,請設置以下變量:
# Specify that GPU direct communication should be used export GMX_ENABLE_DIRECT_GPU_COMM=1 # Specify that GPU PME decomposition should be used export GMX_GPU_PME_DECOMPOSITION=1
通過 -npme <N>標志指定 PME 的總數 GPU 至mdrun。如果沒有 PME GPU 分解,N為 1 ,因為您只能使用單個 PME GPU 。
通過分解,您可以將N設置為正在使用的節點數,以便為每個節點指定一個 PME GPU ,而每個節點中的其他三個 GPU 專用于 PP 。考慮到 PP 和 PME 工作負載的相對計算開銷,這種劃分通常會提供良好的平衡,但建議在任何特定情況下進行實驗。
總結
在這篇文章中,我們描述了我們在增強 GROMACS 多節點可擴展性方面的工作,我們展示了高達 21 倍的性能改進,并詳細介紹了如何使用這些新功能。這使研究人員能夠繼續突破關鍵生物過程知識的界限。
我們繼續通過一系列開發改進 GROMACS ,不僅針對進一步增強的可擴展性,還針對每個計算節點的性能和效率。
GROMACS 2023 的另一個令人興奮的發展是,尖端的 CUDA 圖形技術已被用于提高性能,特別是對于小型科學系統。
按照這些說明,嘗試將您自己的 GROMACS 案例擴展到許多 GPU ,并加速您的科學研究。
你在哪里可以學到更多?加入 GROMACS 論壇。
?