GROMACS是一個用于生物分子系統的模擬軟件包,是世界范圍內使用率最高的科學軟件應用程序之一,也是理解重要生物過程(包括當前新冠病毒大流行的基礎)的關鍵工具。
在以前的職位中,我們展示了最近與核心開發團隊合作進行的優化,這些優化使 GROMACS 在現代多 GPU 服務器上的運行速度比以前快得多。這些優化包括將計算和通信卸載到 GPU ,后者對于那些可以有效地將多個 GPU 并行用于單個模擬的相對較大的情況尤其有利。有關最新異構軟件引擎的并行化和內部工作的更多信息,請參閱GROMACS 中分子動力學模擬的非均勻并行化和加速。
另一個越來越常見的工作流涉及運行許多獨立的 GROMACS 仿真,其中每個仿真都可以相對較小。 NVIDIA GPU 的規模和能力不斷增加,但通常情況下,相對較小的仿真系統的孤立單彈道仿真無法充分利用每個系統上的所有可用計算資源 GPU 。
但是,多部門工作流程可以涉及數十到數千個松散耦合的分子動力學模擬。在此類工作負載中,目標通常不是最小化單個仿真的求解時間,而是最大化整個集成的吞吐量。
按照 GPU 并行運行多個模擬可以顯著提高總體吞吐量,正如之前在GROMACS 中分子動力學模擬的非均勻并行化和加速中針對 GROMACS 所示(圖 11 )。已創建 NVIDIA 多進程服務器( MPS )和多實例 GPU ( MIG )功能,以促進此類工作流,通過使每個 GPU 能夠同時用于多個任務,進一步提高效率。
在這篇文章中,我們演示了為 GROMACS 按照 GPU 運行多個模擬的好處。我們展示了如何利用 MPS ,包括與 MIG 結合使用,在每個 GPU 上并行運行多個模擬,從而實現高達 1 . 8 倍的吞吐量總體改善。
背景資料
硬件
對于本文后面給出的結果,我們在DGX A100服務器上運行,該服務器是 NVIDIA 內部賽琳娜超級計算機的一部分,它具有八個 A100-SXM4-80GB GPU 和兩個 AMD EPYC 7742 (羅馬) 64 核 CPU 插槽。
GROMACS 測試用例
為了生成下一節給出的結果,我們使用 24K 原子核糖核酸酶立方和 96K 原子 ADH-dodec 測試用例,這兩個測試用例都使用 PME 進行遠程電子 CTR 靜態測試。有關更多信息和輸入文件,請參閱GROMACS 中分子動力學模擬的異構并行化和加速補充信息。
我們使用 GROMACS 版本 2021 . 2 (與 CUDA 版本 11 . 2 、驅動程序版本 470 . 57 . 02 一起使用)。對于單個孤立實例(在單個A100-SXM4-80GB GPU 上),使用與后面所述相同的 GROMACS 選項,我們實現了 RNAse 的1083 納秒/天和 ADH 的378 納秒/天。
-update gpu選項對此處的性能至關重要。這將觸發“ GPU – 駐留步驟”,其中每個 timestep 的更新和約束部分被卸載到 GPU 以及默認的“ GPU 強制卸載”行為。如果沒有這一點,在這個硬件上,每種情況下的性能大約降低 2 倍。這對 CPU 和 GPU 可用功能的平衡非常敏感,因此如果在不同的硬件上運行,建議您嘗試使用此選項。
對于本文描述的實驗,我們使用獨立啟動的同一模擬系統的多個實例。這是真實集合模擬的代理,當然,成員模擬和不規則通信之間可能存在細微差異,但仍有以類似方式重疊的范圍。
MPS
隨著 GPU 的規模和功能不斷增加,單個應用程序的執行可能無法充分利用 GPU 提供的所有資源。 NVIDIA 多進程服務( MPS )是一種使多個 CPU 進程提交的計算內核能夠在同一 GPU 上同時執行的工具。這種重疊可能實現更徹底的資源使用和更好的總體吞吐量。
使用 MPS 還可以通過更有效地重疊硬件資源利用率和更好地利用基于 CPU 的并行性,在多個 GPU 之間實現應用程序的強大擴展,如本文后面所述。
2017 年, NVIDIA Volta 架構與 NVIDIA Volta MPS 一起發布,具有增強的功能,包括每個 GPU 最多增加 48 個 MPS 客戶端:這在所有后續 V100 、 Quadro 和 GeForce GPU 上都受支持。
MIG
與 MPS 一樣, NVIDIA 多實例 GPU ( MIG )促進了每個 GPU 的共享,但對資源進行了嚴格的分區。它非常適合在多個不同的用戶之間共享 GPU ,因為每個 MIG 實例都有一組有保證的資源,并且是完全隔離的。
GPU 在選定的 NVIDIA 安培體系結構 MIG 上可用,包括 A100 ,每個 GPU 最多支持七個 MIG 實例。 MIG 可以與 MPS 組合,其中多個 MPS 客戶端可以在每個 MIG 實例上同時運行,每個物理 GPU 最多支持 48 個 MPS 客戶端。
雖然可以跨多個 MIG 實例運行單個應用程序實例,例如使用 MPI , MIG 的目標并不是為該用例提供任何性能改進。 MIG 的主要目的是在每個 GPU 上促進多個不同的應用程序實例。有關更多信息,請參閱通過多實例 GPU 充分利用 NVIDIA A100 GPU 。
績效結果
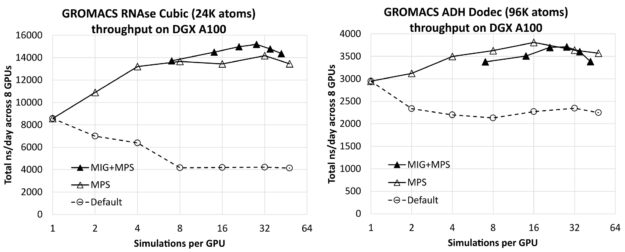
圖 1 顯示了 RNAse (左)和 ADH (右)在 8- GPU DGX A100 服務器上同時運行的所有模擬中,每個 GPU 的模擬次數對總綜合吞吐量(以 ns /天為單位,越高越好)的影響。每種情況下最左邊的結果(每個 GPU 進行一次模擬)僅比相應的單獨模擬結果(前面給出)乘以 8 ( DGX A100 服務器上 GPU 的數量)低幾個百分點。這表明您可以在服務器上有效地運行多個模擬,而不會產生明顯的干擾。
您可以看到,通過增加每個 GPU 的模擬次數,可以獲得實質性的改進,對于相對較小的 RNAse 病例,模擬次數最多為 1 . 8X ,對于較大的 ADH 病例,模擬次數最多為 1 . 3X 。
對于每個測試用例,我們使用每個 GPU 最多七個 MIG 分區,包括沒有 MIG 和有 MIG 的結果。在這種情況下,MPS用于每個 GPU 運行多個進程,每個物理 GPU 最多運行48個MPS客戶端,其中 MIG 情況下的最大客戶端總數為42:7個 MIG 分區中的每個分區有6個MPS客戶端。作為參考,我們還包括了沒有MPS或 MIG 的默認結果,對于這些結果,我們認為按照 GPU 運行多個模擬沒有任何好處。
對于每個測試用例,最左邊的 MIG+MPS 結果是每個 GPU 進行 7 次模擬:每個 MIG 客戶端進行一次模擬(即,沒有 MPS)。我們發現這些“純 MIG”性能結果與相應的“純 MPS”結果相比沒有優勢。對于 RNAse,純 MIG 類似于純 MPS,而對于 ADH 則低于純 MPS。然而,將 MIG 與 MPS 相結合會導致 RNAse 的最佳整體結果,比最佳純 MPS 結果高約 7%。它導致性能與 ADH 的純 MPS 相當,但略低于純 MPS。
對于 RNAse ,性能最好的配置是每個 MIG 四個 MPS 客戶端,即每個 GPU 總共 28 個模擬。對于 ADH ,最好的配置是使用純 MPS ,每 GPU 進行 16 次模擬,而不使用 MIG 。
當 MIG 處于活動狀態時,它強制將每個模擬隔離到 GPU 硬件的特定分區,這可能是有利的,具體取決于測試用例關鍵路徑的特定數據訪問模式。另一方面,在沒有 MIG 的情況下,每個仿真都可以以更動態的方式訪問 GPU 上的資源,這也是有利的。
MIG 的好處可能取決于測試用例,正如我們前面討論的那樣。令人欣慰的是,MPS在有 MIG 和沒有 MIG 的情況下都是有效的,特別是在有些用例中 MIG 出于其他原因是可取的,例如在用戶之間共享 GPU 。
GROMACS 是異構的,并且具有靈活性,在這方面,計算可以卸載到 GPU ,其中 CPU 可以同時使用。我們的運行使用了力計算的默認選項,將非粘結力和 PME 力計算映射到 GPU ,同時使用 CPU 進行粘結力計算。這種配置通常會導致資源的良好重疊使用。
我們嘗試將粘結力計算卸載到 GPU (使用 -bonding GPU 選項),性能類似,但在所有情況下都略低。如前所述,我們使用 GPU – 駐留步驟。我們嘗試將更新和約束映射到 CPU ,我們還觀察到了根據 GPU 運行多個模擬的好處。
對于較大的 ADH 情況,可實現的吞吐量大大低于將更新和約束卸載到 GPU 時的吞吐量。然而,對于較小的 RNAse 情況,盡管在每 GPU 運行一次(或幾次)模擬時吞吐量較低,但在每 GPU 運行八次或更多模擬時,無論是否卸載此部分,我們都看到了類似的吞吐量。行為可能因測試用例和硬件而異,因此最好使用所有可用的運行時組合進行實驗。
我們還在不同的硬件上重復了核糖核酸酶的MPS實驗:NVIDIA A40和V100-SXM2 GPU ,我們還發現,每個 GPU 運行多個模擬可以提高吞吐量,盡管程度低于A100。鑒于這些 GPU 的規格相對較低,這并不奇怪。A40為1.5倍,V100-SXM2為1.4倍,觀察到的吞吐量改善明顯低于A100的1.8倍,但仍然值得。
這些結果表明,通過在 MPS 中按 GPU 運行多個進程,并將 MIG 與 MPS 相結合,可以實現大的吞吐量改進。最佳配置(包括 GROMACS 中的計算卸載選項)取決于具體情況,我們再次建議進行實驗。以下各節描述了這些模擬是如何編排的。
運行配置詳細信息
在本節中,我們將提供用于生成結果的腳本,并描述其中包含的命令,作為您自己工作流的參考或起點。
純議員選舉
以下腳本使用 MPS 向 8- GPU DGX A100 服務器啟動多個模擬。
1 #!/bin/bash
2 # Demonstrator script to run multiple simulations per GPU with MPS on DGX-A100
3 #
4 # Alan Gray, NVIDIA
5
6 # Location of GROMACS binary
7 GMX=/lustre/fsw/devtech/hpc-devtech/alang/gromacs-binaries/v2021.2_tmpi_cuda11.2/bin/gmx
8 # Location of input file
9 INPUT=/lustre/fsw/devtech/hpc-devtech/alang/Gromacs_input/rnase.tpr
10
11 NGPU=8 # Number of GPUs in server
12 NCORE=128 # Number of CPU cores in server
13
14 NSIMPERGPU=16 # Number of simulations to run per GPU (with MPS)
15
16 # Number of threads per simulation
17 NTHREAD=$(($NCORE/($NGPU*$NSIMPERGPU)))
18 if [ $NTHREAD -eq 0 ]
19 then
20 NTHREAD=1
21 fi
22 export OMP_NUM_THREADS=$NTHREAD
23
24 # Start MPS daemon
25 nvidia-cuda-mps-control -d
26
27 # Loop over number of GPUs in server
28 for (( i=0; i<$NGPU; i++ ));
29 do
30 # Set a CPU NUMA specific to GPU in use with best affinity (specific to DGX-A100)
31 case $i in
32 0)NUMA=3;;
33 1)NUMA=2;;
34 2)NUMA=1;;
35 3)NUMA=0;;
36 4)NUMA=7;;
37 5)NUMA=6;;
38 6)NUMA=5;;
39 7)NUMA=4;;
40 esac
41
42 # Loop over number of simulations per GPU
43 for (( j=0; j<$NSIMPERGPU; j++ ));
44 do
45 # Create a unique identifier for this simulation to use as a working directory
46 id=gpu${i}_sim${j}
47 rm -rf $id
48 mkdir -p $id
49 cd $id
50
51 ln -s $INPUT topol.tpr
52
53 # Launch GROMACS in the background on the desired resources
54 echo "Launching simulation $j on GPU $i with $NTHREAD CPU thread(s) on NUMA region $NUMA"
55 CUDA_VISIBLE_DEVICES=$i numactl --cpunodebind=$NUMA $GMX mdrun \
56 -update gpu -nsteps 10000000 -maxh 0.2 -resethway -nstlist 100 \
57 > mdrun.log 2>&1 &
58 cd ..
59 done
60 done
61 echo "Waiting for simulations to complete..."
62 wait
- 第 7 行和第 9 行分別指定 GROMACS 二進制文件和測試用例輸入文件的位置。
- 第 11-12 行指定服務器的固定硬件詳細信息,該服務器有 8 個 GPU 和 128 個 CPU 內核。
- 第 14 行指定了每個 GPU 的模擬次數,可以改變模擬次數以評估性能,正如生成上述結果所做的那樣。
- 第 17-21 行計算每個模擬應分配多少 CPU 個線程。
- 第 25 行啟動 MPS 守護進程,使從單獨模擬啟動的內核能夠在同一 GPU 上同時執行。
- 第 28-40 行在服務器中的 GPU 上循環,并為每個特定 GPU 分配一組適當的 CPU 內核(“NUMA區域”)。此映射特定于DGX A100拓撲,該拓撲具有兩個AMD CPU s,每個AMD CPU 具有四個NUMA區域。我們安排特定的編號以獲得最佳親和力。有關更多信息,請參見DGX A100 用戶指南中的第1.3節。
- 第 43-49 行循環每個 GPU 的模擬次數,并創建模擬特有的工作目錄。
- 第 51 行在此唯一工作目錄中創建指向輸入文件的鏈接。
- 第 55-57 行啟動每個模擬,使用
CUDA_VISIBLE_DEVICES環境變量將其限制為所需的 GPU ,使用 numactl 實用程序將其限制為所需的 CPU NUMA 區域。可以使用< code > apt install numactl </ code >提前安裝該實用程序。
-update GPU 選項與默認的 GPU 強制卸載行為相結合,對性能至關重要(見上文),而-nsteps 10000000 -maxh 0.2 -resethway組合的結果是運行每個模擬 12 分鐘( 0 . 2 小時),其中內部計時器會在中途重置,以消除任何初始化開銷。-nstlist 100指定 GROMACS 應每隔 100 步重新生成內部鄰居列表,其中這是一個可調參數,影響性能但不影響正確性。)
結合 MIG 和 MPS 運行
以下腳本是前一個腳本的版本,擴展為支持每個 GPU 的多個 MIG 實例,其中每個 MIG 實例可以使用 MPS 啟動多個仿真。
1 #!/bin/bash
2 # Demonstrator script to run multiple simulations per GPU with MIG+MPS on DGX-A100
3 #
4 # Alan Gray, NVIDIA
5
6 # Location of GROMACS binary
7 GMX=/lustre/fsw/devtech/hpc-devtech/alang/gromacs-binaries/v2021.2_tmpi_cuda11.2/bin/gmx
8 # Location of input file
9 INPUT=/lustre/fsw/devtech/hpc-devtech/alang/Gromacs_input/adhd.tpr
10
11 NGPU=8 # Number of GPUs in server
12 NCORE=128 # Number of CPU cores in server
13
14 NMIGPERGPU=7 # Number of MIGs per GPU
15 NSIMPERMIG=3 # Number of simulations to run per MIG (with MPS)
16
17 # Number of threads per simulation
18 NTHREAD=$(($NCORE/($NGPU*$NMIGPERGPU*$NSIMPERMIG)))
19 if [ $NTHREAD -eq 0 ]
20 then
21 NTHREAD=1
22 fi
23 export OMP_NUM_THREADS=$NTHREAD
24
25 # Loop over number of GPUs in server
26 for (( i=0; i<$NGPU; i++ ));
27 do
28 # Set a CPU NUMA specific to GPU in use with best affinity (specific to DGX-A100)
29 case $i in
30 0)NUMA=3;;
31 1)NUMA=2;;
32 2)NUMA=1;;
33 3)NUMA=0;;
34 4)NUMA=7;;
35 5)NUMA=6;;
36 6)NUMA=5;;
37 7)NUMA=4;;
38 esac
39
40 # Discover list of MIGs that exist on this GPU
41 MIGS=`nvidia-smi -L | grep -A $(($NMIGPERGPU+1)) "GPU $i" | grep MIG | awk '{ print $6 }' | sed 's/)//g'`
42 MIGARRAY=($MIGS)
43
44 # Loop over number of MIGs per GPU
45 for (( j=0; j<$NMIGPERGPU; j++ ));
46 do
47
48 MIGID=${MIGARRAY[j]}
49 # Start MPS daemon unique to MIG
50 export CUDA_MPS_PIPE_DIRECTORY=/tmp/$MIGID
51 mkdir -p $CUDA_MPS_PIPE_DIRECTORY
52 CUDA_VISIBLE_DEVICES=$MIGID nvidia-cuda-mps-control -d
53
54 # Loop over number of simulations per MIG
55 for (( k=0; k<$NSIMPERMIG; k++ ));
56 do
57
58 # Create a unique identifier for this simulation to use as a working directory
59 id=gpu${i}_mig${j}_sim${k}
60 rm -rf $id
61 mkdir -p $id
62 cd $id
63
64 ln -s $INPUT topol.tpr
65
66 # Launch GROMACS in the background on the desired resources
67 echo "Launching simulation $k on MIG $j, GPU $i with $NTHREAD CPU thread(s) on NUMA region $NUMA"
68 CUDA_VISIBLE_DEVICES=$MIGID numactl --cpunodebind=$NUMA $GMX mdrun \
69 -update gpu -nsteps 10000000 -maxh 0.2 -resethway -nstlist 100 \
70 > mdrun.log 2>&1 &
71 cd ..
72 done
73 done
74 done
75 echo "Waiting for simulations to complete..."
76 wait
與純 MPS 腳本的主要區別以粗體突出顯示:
- 第 14 行指定每個 GPU 的 MIG 實例數,設置為最多 7 個。使用以下命令提前在八個 GPU 中的每一個上創建實例:
for gpu in 0 1 2 3 4 5 6 7
do
nvidia-smi mig -i $gpu --create-gpu-instance \
1g.10gb,1g.10gb,1g.10gb,1g.10gb,1g.10gb,1g.10gb,1g.10gb \
--default-compute-instance
done
- 第 15 行指定每個 MIG 實例要運行的模擬數量。
- 第 18 行調整 CPU 核心分配計算,以正確地考慮每個 GPU 的總模擬次數。
- 第 41-42 行位于循環(物理) GPU s 內,列出與 GPU 關聯的 MIG s 唯一的 ID ,并創建包含這七個 ID 的可索引數組。
- 第 45 行向循環嵌套添加一個新的中間級別,對應于每個 GPU 的多個 MIG s 。
- 第 48-52 行根據 MIG 啟動一個單獨的 MPS 守護程序,這是結合 MIG 和 MPS 的要求。
- 第 55 行在每個 MIG 的模擬次數上循環。它繼續像以前一樣啟動每個模擬,但現在每個模擬都被限制為使用唯一 MIG ID 的特定 MIG 實例。
多流程的其他優勢
到目前為止,我們已經向您展示了如何按照 GPU 運行多個進程可以為 GROMACS 帶來巨大的好處,并且我們提供了具體的示例來演示。類似的技術可以在更廣泛的用例和場景中提供好處,包括 GROMACS 和其他應用程序。在本節中,我們將簡要討論其中的一些。
GROMACS 多模擬框架
在這篇文章中,我們使用 shell 腳本中的循環并行啟動了多個模擬。內置的 GROMACS 多仿真框架提供了一種替代機制,其中通過-multidir選擇mdrun啟動 GROMACS 時,將多個 MPI 任務映射到多個仿真。以類似的方式,最大化吞吐量的好處也可以應用于該機制。
為了評估這一點,我們沒有直接使用 MPI 啟動 GROMACS ,而是通過一個包裝器腳本啟動 GROMACS ,該腳本可以為每個 MPI 級別設置適當的環境變量和numactl選項。可以使用環境變量發現秩,例如 OpenMPI 的OMPI_COMM_WORLD_LOCAL_RANK。另一種方法是以類似的方式使用 MPI 啟動器的綁定功能。
我們的實驗表明, MPS 的行為與前面描述的一樣。然而,相比之下,我們并沒有看到 MPS 和 MIG 結合的任何額外好處,這需要進一步的研究。
多模擬框架還支持在集合內的模擬之間進行不頻繁的副本交換,使用-replex選擇mdrun或AWH 多步行機方法在一個模擬中應用多個獨立的偏置電位。對于這些工作流,性能行為取決于用例的特定細節,因此我們建議進行實驗。有關-replex選項的更多信息,請參閱副本交換模擬簡介教程。
用于多重 GPU 強擴展的 MPS
這篇文章探討了 MPS (和 MIG )的好處,以提高并行運行的許多獨立模擬的吞吐量。另一個共同目標是通過并行使用多個 GPU 來最小化單個模擬的求解時間。
通常,單獨的 CPU 任務(如 MPI 任務)用于控制每個 GPU ,并執行未卸載到 GPU 的任何計算工作負載。在某些情況下,每個 GPU 運行多個 CPU 任務是有益的,因為這可以為重疊的 CPU 計算 GPU 計算和通信提供額外的機會。按照 GPU 運行多個進程也有助于重新平衡并行運行中固有的負載不平衡。有關更多信息,請參閱本節后面討論的 GROMACS 示例。
此方法還可以增加 CPU 上基于任務的并行程度,并增強應用程序中任何 CPU 駐留的并行工作負載的性能。當 MPS 處于活動狀態時,與多個任務關聯的多個內核可以在每個 GPU 上并行執行。最好是進行實驗,以發現它是否能對特定情況有益,并找到最佳配置。
格羅馬克的例子
下面是一個具體的例子。在以前的職位中,我們將重點放在每個 GROMACS 模擬并行運行四個 GPU 的情況下,以最小化求解時間。我們展示了這四個 GPU 如何有效地平衡三個 PP 任務和一個 PME 任務。
但是,如果您試圖將該配置調整為兩個 GPU ,最自然的方法是分配一個 PP GPU 和一個 PME GPU 。這不會產生良好的性能,因為 PP GPU 的工作量要大得多。
最好將四 GPU 配置映射到兩 GPU 配置,并激活 MPS 以啟用內核重疊。 GPU 中的一個重疊兩個 PP 任務,而另一個重疊兩個 PP 和一個 PME 。這將導致更好的負載平衡和更快的解決時間。在GROMACS 中分子動力學模擬的非均勻并行化和加速論文中,該技術用于生成圖 12 的兩個 GPU 強縮放結果。同樣,我們建議對任何特定情況進行實驗。
與計算重疊的 I / O
這篇文章展示了按照 GPU 運行多個進程如何通過重疊提供好處。好處不僅限于計算和通信。此解決方案也適用于花費大量 I / O 時間的情況。在這些情況下,一個實例的 I / O 組件可以與另一個實例的計算組件重疊,以提高總體吞吐量。 MPS 使內核能夠與其他計算內核、通信或文件 I / O 并行執行。
如果您有問題或建議,請在下面進行評論。
?